Toronto, Ontario--(Newsfile Corp. - May 4, 2026) - Theralase® Technologies Inc. (TSXV: TLT) (OTCQB: TLTFF) ("Theralase®" or the "Company"), a clinical stage pharmaceutical company dedicated to the research and development of energy-activated small molecules for the safe and effective destruction of various cancer, bacteria and viruses has released the Company's audited consolidated financial statements for the twelve-month period ended December 31st, 2025 ("Financial Statements").
Theralase® will be hosting a conference call on May 12th at 11:00 am ET, which will include a presentation of the financial and operational results for the fiscal year ending December 31st, 2025.
To ensure Theralase® has time to address questions during the call, please e-mail them in advance to mperraton@theralase.com.
| Zoom Meeting Link: | https://us02web.zoom.us/j/82405815948 |
| Conference Call in: | 1-647-558-0588 (Canada) / 1-646-558-8656 (US) - not required for those attending by Zoom. |
An archived version will be available on the website following the conference call.
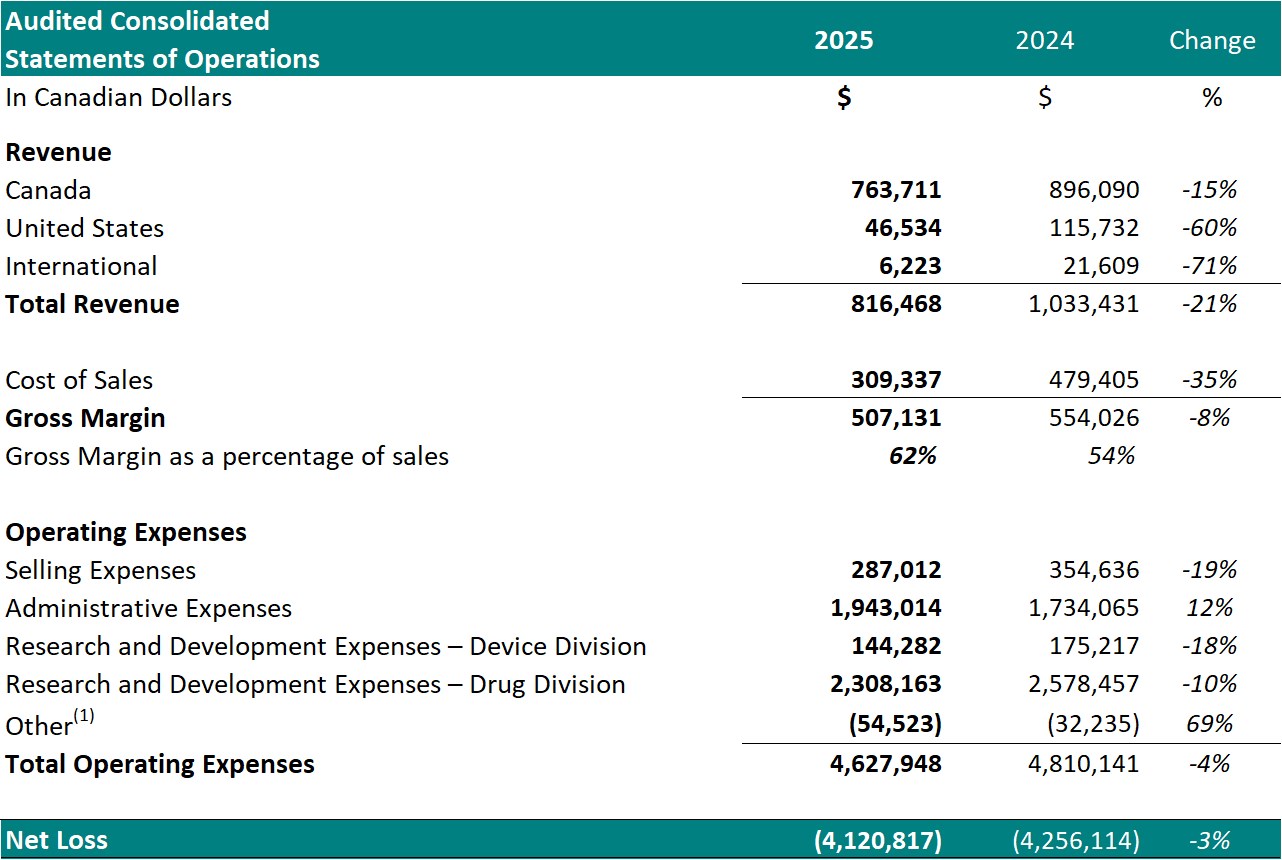
Table 1: Financial Summary for the year ended December 31st
To view an enhanced version of this graphic, please visit:
https://images.newsfilecorp.com/files/2786/295879_0ea03f92e40a1b12_001full.jpg
Financial Highlights:
For the year ended December 31st, 2025 (All funds in Canadian Dollars):
- Total revenue decreased to $816,468 from $1,033,431 for the same period in 2024, a 21% decrease.
Cost of sales for the year ended December 31, 2025, was $309,337 (38% of revenue) resulting in a gross margin of $507,131 (62% of revenue). In comparison, the cost of sales for the same period in 2024 was $479,405 (46% of revenue) resulting in a gross margin of $554,026 (54% of revenue).
- Selling expenses decreased to $287,012 from $354,636 for the same period in 2024, a 19% decrease. The decrease in selling expenses is primarily a result of decreased spending on salaries (17%) and commissions (38%).
- Administrative expenses for year ended December 31, 2025, increased to $1,943,014 from $1,734,065 for the same period in 2024, a 12% increase. The increase in administrative expenses is primarily a result of increased spending on general and administrative expenses (17%) and professional fees (70%).
- Net research and development expenses for the year ended December 31, 2025, decreased to $2,452,445 from $2,753,674 for the same period in 2024, a 9% decrease. The decrease in research and development expenses is attributed to a decrease in costs for Study II patient enrollment and treatment. Research and development expenses represented 53% of the Company's operating expenses and represent investment into the research and development of the Company's Drug Division.
- The net loss for the year ended December 31, 2025, was $4,120,817, which included $840,227 of net non-cash expenses (i.e.: amortization, stock-based compensation expense and foreign exchange gain/loss). This compared to a net loss for the same period in 2024 of $4,256,114, which included $793,181 of net non-cash expenses. The Drug Division represented $3,400,303 (83%) of this loss. The decrease in net loss is primarily attributed to decreased spending on research and development expenses in Study II.
Operational Highlights:
Collaborative Clinical Development Agreement
On January 12, 2026, the Company announced that it had entered into a collaborative clinical development agreement dated January 9, 2026 with Ferring Pharmaceuticals, expanding the Company's existing Phase II NMIBC clinical program (NCT03945162) through the addition of a new cohort evaluating Ruvidar® (TLD-1433) in combination with Adstiladrin® (nadofaragene firadenovec-vncg) for adult patients diagnosed with high-risk Bacillus Calmette-Guérin ("BCG")-Unresponsive Non-Muscle Invasive Bladder Cancer ("NMIBC") Carcinoma In-Situ ("CIS") with or without papillary disease (±Ta/T1) ("Study II"). Under the terms of the agreement, the Company will remain the sponsor of the study, with both parties providing clinical oversight through a joint development committee. The new cohort is expected to be enrolled and treated initially in the United States and, subject to written agreement, may expand into Canada or other jurisdictions.
Study II Interim Clinical Data
Cohort 1
Theralase® has successfully completed its targeted enrollment for Study II, exceeding expectations with 91 patients treated by Clinical Study Sites ("CSSs") and may continue to enroll additional patients as remaining sites finalize and close to enrollment.
According to the clinical study design, a patient is considered to have completed Study II, if they received the Study Procedure and have been assessed by the Principal Investigator ("PI") for up to 15 months or upon earlier withdrawal by the PI due to non-response or non-compliance.
Based on this definition, 82 patients have completed Study II, while 9 remain active with pending clinical data. Interim results supporting the Study II endpoints are summarized below.
A total of 91 patients have been enrolled and treated. The cohort was predominantly ≥65 years of age (81%), male (81%), and white (83%). Tumour staging was distributed as follows: 81% carcinoma in situ ("CIS") only, 12% CIS + T1, and 7% CIS + Ta. In addition, 98% of patients were classified as BCG-unresponsive and 2% as BCG-intolerant. The median number of prior BCG instillations was 15.5.
As of April 30, 2026, 89 patients have been assessed for response outcomes and are evaluable for the primary endpoint analysis.
Primary Endpoint Performance (Complete Response at any Point in Time)
The primary endpoint of Study II is the achievement of Complete Response ("CR") at any point in time following administration of the Study Procedure. Interim analysis demonstrates that 65.2% (58 out of 89) evaluable patients achieved CR.
| Primary Endpoint Performance (CR at any Point in Time) | |||
| # | % | Confidence Interval (95%) | |
| Complete Response ("CR") | 58/89 | 65.2% | [49.4, 80.9] |
| Total Response (CR and IR) | 65/89 | 73.0% | [56.4, 89.7] |
Table 2: Primary Endpoint Performance
Approximately, 2 out of 3 patients diagnosed with BCG-Unresponsive NMIBC CIS (with or without Ta/T1) achieved a CR following treatment with the Theralase® Study Procedure.
Secondary Endpoint Performance (Duration of CR - 12 Months)
The secondary endpoint evaluates the sustainability of CR at 12 months, after initial CR determination (450 days post-treatment). Among patients evaluable for durability of response, 40.4% (21 of 52 evaluable patients) maintained a CR at 450 days.
| Secondary Endpoint Performance (Duration of CR) (450 Days) | |||
| # | % | Confidence Interval (95%) | |
| Complete Response (CR) | 21/52 | 40.4% | [24.0, 56.7] |
| Total Response (CR and IR) | 22/52 | 42.3% | [26.5, 58.1] |
Table 3: Secondary Endpoint Performance
Tertiary Endpoint Performance (Safety)
The tertiary endpoint is defined as patients who are diagnosed with a Serious Adverse Event ("SAE") ≥ 4 directly caused by the Study Drug or Study Device, which did not resolve within 450 days.
Theralase® and the independent Data Safety Monitoring Board believe all SAEs reported to date are unrelated or unlikely related to the Study Drug or Study Device.
The tertiary endpoint assesses the safety profile of the Study Procedure.
| Note: | A SAE is defined as any untoward medical occurrence that at any dose: Is serious or life-threatening, requires inpatient hospitalization or prolongation of existing hospitalization, results in persistent or significant disability/incapacity, is a congenital anomaly/birth defect or results in death. |
Treatment Emergent Adverse Events ("TEAEs") were noted, but did not meet the SAE criteria. TEAEs included urinary frequency (65%), hematuria (62.5%) and urinary urgency (53.8%), which resolved within 1 month of treatment.
There have been 24 SAEs reported: 1 x Grade I, 3 x Grade II, 13 x Grade III, 5 x Grade IV (all resolved between 1 to 82 days) and 2 x Grade V. A high majority of SAEs were not treatment related and none were directly related to the Study Drug or Study Device.
| Tertiary Endpoint Performance (Safety) (450 Days) | ||
| # | % | |
| Safety | 82/82 | 100.0% |
Table 4: Tertiary Endpoint Performance
Duration of CR - Extended Time Points
Patients who have completed the study are followed for up to 3 years after initial treatment at extended time points.
| Duration of CR | |||
| Time | # | % | Confidence Interval (95%) |
| 2 Years | 10/52 | 19.2% | [7.9, 30.5] |
| 3 Years | 10/52 | 19.2% | [7.9, 30.5] |
Table 5: Duration of CR at Extended Time Points
One patient demonstrated CR for 7 years, after one Study Procedure.
Based on Kaplan-Meier analysis, if CR is obtained, the long term estimated probability of remaining cancer free at 1, 2 and 3 years is 48.6%, 34.5% and 25.4%, respectively.
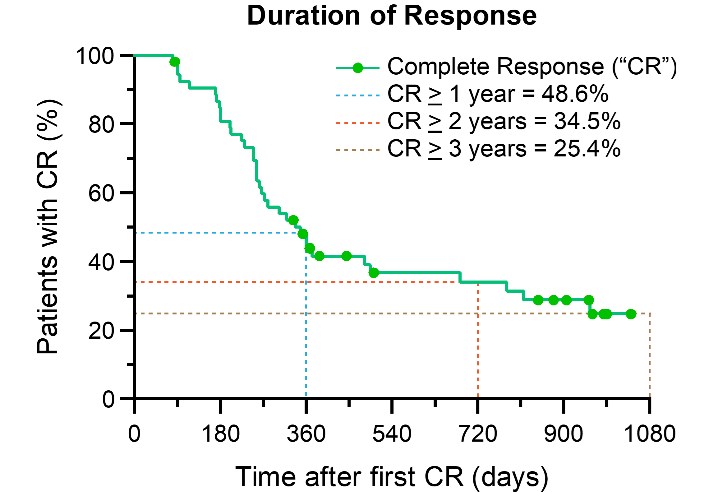
Figure 1: Kaplan-Meier Curve
To view an enhanced version of this graphic, please visit:
https://images.newsfilecorp.com/files/2786/295879_0ea03f92e40a1b12_002full.jpg
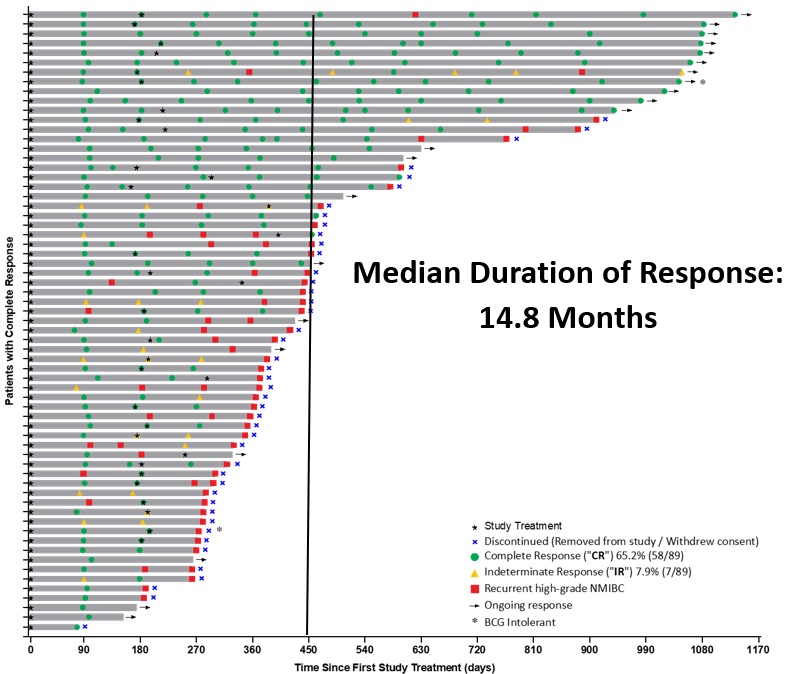
Figure 2: Swimmer's Plot
To view an enhanced version of this graphic, please visit:
https://images.newsfilecorp.com/files/2786/295879_figure2.jpg
| Note: | These clinical results are interim in nature. Study II remains ongoing. Additional clinical data may influence or alter current response trends. |
Regulatory Pathway, Commercialization Strategy and FDA Guidance
If approved by Health Canada and the FDA, the clinical data collected from Study II represents a transformative therapeutic option for patients diagnosed with BCG-Unresponsive NMIBC CIS, who would otherwise face radical cystectomy (surgical removal of the bladder). The Theralase® Study Procedure has demonstrated a robust CR and sustained durability of that response, with the majority of patients receiving only a single Study Procedure.
Following the completion of patient follow-up and final clinical data analysis, Theralase® intends to submit a New Drug Submission ("NDS") to Health Canada and a New Drug Application ("NDA") to the United States Food and Drug Administration ("FDA") in 3Q2026, under a rolling review, with regulatory decisions anticipated in 1H2027.
Cohort 2
Theralase®, in conjunction with Ferring Pharmaceutical, subject to FDA approval, is preparing to launch a combinational clinical study to investigate the safety and efficacy of combining light-activated Ruvidar® with Adstiladrin.
It is anticipated that the complementary mechanisms of action (Ruvidar® targets bladder cancer cells directly, while Adstiladrin® targets health bladder cells to produce Interferon to stimulate the innate and adaptive immune system) will provide an additive effect in the treatment of patients being treated for BCG-Unresponsive NMIBC CIS.
In the Study Procedure, patients will be treated with Ruvidar® (1 hour of drug instillation, 1 hour of light activation), then at another visit, they will be instilled with Adstiladrin® (1 hour procedure), both in outpatient procedures. Under the clinical protocol, the patient may receive up to 4 treatments of Adstiladrin®.
The presiding uro-oncologist will have the option to deliver an additional re-induction Study Procedure, if the patient recurs.
The patient will be followed for 15 months after initial Study Procedure and up to 3 years for post-study follow-up.
Commercialization and Strategic Partnerships
In parallel with the finalization of Study II, Theralase® is actively pursuing commercialization opportunities and strategic partnerships to support the global marketing and distribution of Ruvidar®. The Company is pursuing discussions with pharmaceutical companies across multiple geographic regions regarding:
- Licensing arrangements for Ruvidar® in the treatment of BCG-Unresponsive NMIBC CIS in various geographic territories
- Collaborative clinical research initiatives focused on the application of light-activated Ruvidar® for broader NMIBC indications
- Collaborative clinical research combining Ruvidar®, with other FDA-approved drugs to enhance treatment efficacy
About Study II:
Study II utilizes the therapeutic dose of the patented drug, Ruvidar® (TLD-1433) activated by the patented study device, the TLC-3200 Medical Laser System. Study II has enrolled and treated 91 BCG-Unresponsive NMIBC CIS patients in 11 clinical study sites located in Canada and the United States.
About Theralase® Technologies Inc.:
Theralase® is a clinical stage pharmaceutical company dedicated to the research and development of energy-activated small molecules for the safe and effective destruction of cancer, bacteria and viruses.
Additional information is available at www.theralase.com and www.sedarplus.ca
Neither TSX Venture Exchange nor its Regulation Services Provider (as that term is defined in the policies of the TSX Venture Exchange) accepts responsibility for the adequacy or accuracy of this release.
Forward Looking Statements
This news release contains Forward-Looking Statements ("FLS") within the meaning of applicable Canadian securities laws. Such statements include; but, are not limited to statements regarding the Company's proposed development plans with respect to small molecules and their drug formulations. FLS may be identified by the use of the words "may, "should", "will", "anticipates", "believes", "plans", "expects", "estimate", "potential for" and similar expressions; including, statements related to the current expectations of the Company's management regarding future research, development and commercialization of the Company's small molecules; their drug formulations; preclinical research; clinical studies and regulatory approvals.
These statements involve significant risks, uncertainties and assumptions; including, the ability of the Company to fund and secure regulatory approvals to successfully complete various clinical studies in a timely fashion and implement its development plans. Other risks include: the ability of the Company to successfully commercialize its small molecule and drug formulations; access to sufficient capital to fund the Company's operations is available on terms that are commercially favorable to the Company or at all; the Company's small molecule and formulations may not be effective against the diseases tested in its clinical studies; the Company fails to comply with the terms of license agreements with third parties and as a result loses the right to use key intellectual property in its business; the Company's ability to protect its intellectual property; the timing and success of submission, acceptance and approval of regulatory filings. Many of these factors that will determine actual results are beyond the Company's ability to control or predict.
Readers should not unduly rely on these FLS, which are not a guarantee of future performance. There can be no assurance that FLS will prove to be accurate as such FLS involve known and unknown risks, uncertainties and other factors which may cause actual results or future events to differ materially from the FLS.
Although the FLS contained in the press release are based upon what management currently believes to be reasonable assumptions, the Company cannot assure prospective investors that actual results, performance or achievements will be consistent with these FLS.
All FLS are made as of the date hereof and are subject to change. Except as required by law, the Company assumes no obligation to update such FLS.
For investor information on the Company, please feel to reach out Investor Inquiries - Theralase Technologies.
For More Information:
1.866.THE.LASE (843.5273)
416.699.LASE (5273)
www.theralase.com
Kristina Hachey, CPA
Chief Financial Officer X 224
khachey@theralase.com
![]()
To view the source version of this press release, please visit https://www.newsfilecorp.com/release/295879
Source: Theralase Technologies Inc.